About Pairoscope
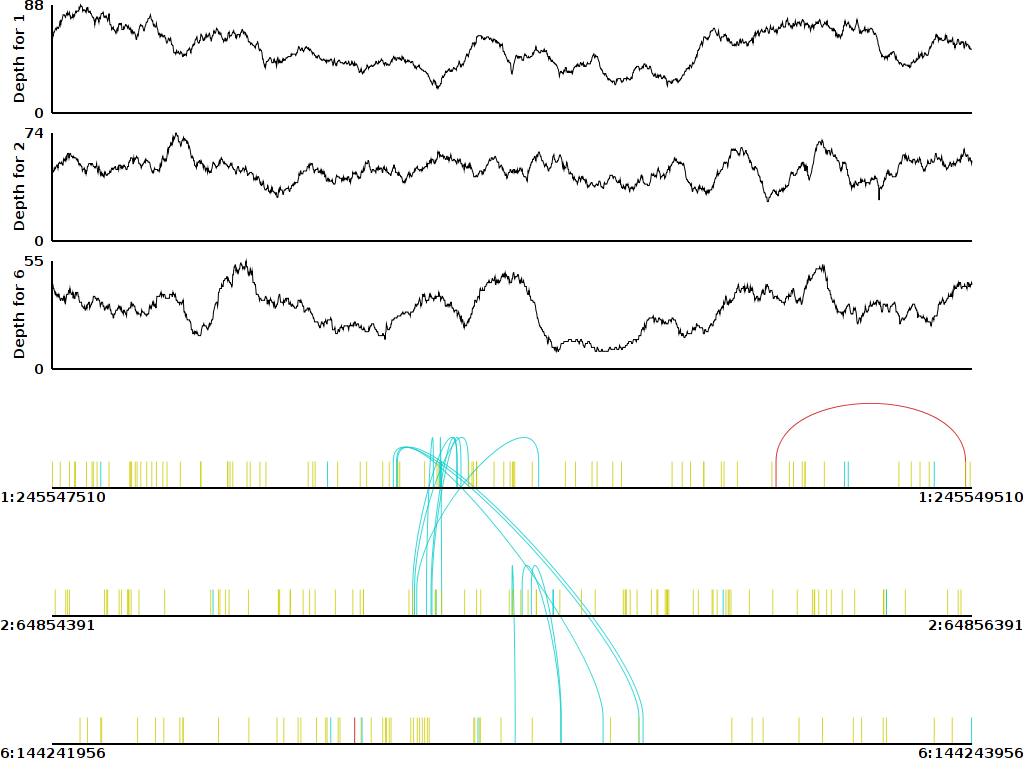
Pairoscope (formerly Yenta) was developed as a quick and simple way to generate diagrams indicating the relationship of paired end sequencing reads. It functions by displaying multiple genomic regions, their read depth at each base in the region and arcs within or between regions to indicate pairing information. Currently, Pairoscope supports the graphing of multiple regions, but they must be at different locations: the behavior of Pairoscope is undefined if attempting to graph the same region twice. It uses a bam file as input, and color codes the read pair graph to reflect different types of abnormally oriented reads. Individual reads are represented as vertical lines drawn at the genomic location where the read mapped. When both reads of a pair are present in the displayed region, the reads are linked by an arc. Reads lying at the same position are drawn on top of each other. In addition to displaying read pair information, Pairoscope also displays the read depth over the region as a separate graph. At the moment, Pairoscope only provides display of reads with abnormal orientations with respect to the reference; deletion and insertion events are not yet supported. In addition, it assumes orientations and colors reflecting the expected orientations for a standard Illumina Paired-End library. It is in beta right now and stability may be an issue.